Intrinsically disordered proteins and regions (IDP/IDRs) are widespread in living organisms, and perform various essential molecular functions. These functions are summarized as six general categories, including entropic chain, assembler, scavenger, effector, display site, and chaperone. The alteration of IDR functions is responsible for many human diseases. Computational function prediction of disordered proteins is helpful for the studies of drug target discovery and rational drug design. Current proposed computational methods mainly focus on predicting the entropic chain function of IDRs, while the computational predictive methods for the remaining five molecular functions of IDRs are desired. Motivated by the growing numbers of experimental annotated functional sequences and the need to expand the coverage of disordered protein function predictors, we proposed DMFpred for disordered molecular functions prediction, covering disordered assembler, scavenger, effector, display site and chaperone.
DMFpred employs the Protein Cubic Language Model (PCLM), which incorporates three protein language models for characterizing sequences, structural and functional features of proteins, and attention-based alignment for understanding the relationship among three captured features and generating a joint representation of proteins (see Fig.1A). The PCLM was pre-trained with large-scaled IDR sequences and fine-tuned with functional annotation sequences for molecular function prediction (see Fig.1B).
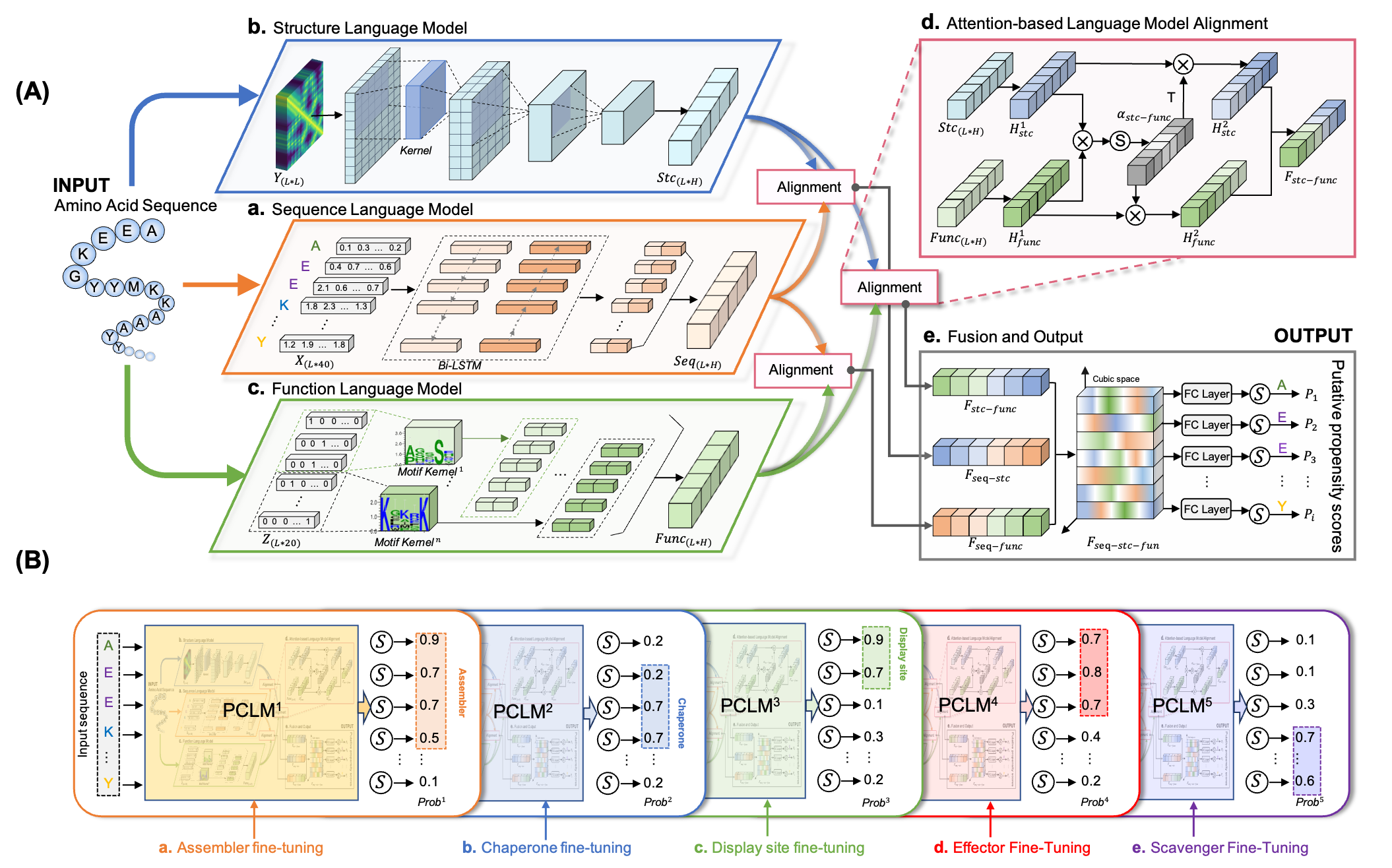
Figure.1 The flowchart of DMFpred. (A) Architecture of the protein cubic language model (PCLM). (B)The PCLM's functional specific fine-tuning for predictiong binding related molecular functions of intrinsically disordered proteins.
Datasets and Supporting Information used in this study can be downloaded here:
· Molecular function benchmark dataset:
· Intrinsically disordered pre-training dataset:
· ELM motifs:
We acknowledge with thanks the following software used in this server:
PSI-BLAST: The protein sequence similarity search.
HH-suite3: Protein sequence alignment based on hidden Markov models (HMMs).
CCMpred: The residue-residue contacts generation of proteins.
FIMO: The Motifs searching tools.
Upon the usage the users are requested to use the following citation:
· Pang, Y. and Liu, B. DMFpred: predicting protein disorder molecular functions based on protein cubic language model. PLOS Computational Biology, 2022, 18(10): e1010668.