Intrinsically disordered regions and proteins (IDPs/IDRs) are functionally important regions and proteins that exist as highly dynamic conformations under natural physiological conditions. IDPs/IDRs exhibit a broad range of molecular functions, and their functions involve binding interactions with partners and remaining native structural flexibility. The rapid increase in the number of proteins in sequence databases and the diversity of disordered functions challenge existing computational methods for predicting protein intrinsic disorder and disordered functions. A disordered region interacts with different partners to perform multiple functions, and these disordered functions exhibit different dependencies and correlations. In this study, we introduce DisoFLAG, a computational method that leverages a graph-based interaction protein language model (GiPLM) for jointly predicting disorder and its multiple potential functions. GiPLM integrates protein semantic information based on pre-trained protein language models into graph-based interaction units to enhance the correlation of the semantic representation of multiple disordered functions. The DisoFLAG predictor takes amino acid sequences as the only inputs and provides predictions of intrinsic disorder and six disordered functions for proteins, including protein-binding, DNA-binding, RNA-binding, ion-binding, lipid-binding, and flexible linker. We evaluated the predictive performance of DisoFLAG following the Critical Assessment of protein Intrinsic Disorder (CAID) experiments, and the results demonstrated that DisoFLAG offers accurate and comprehensive predictions of disordered functions, extending the current coverage of computationally predicted disordered function categories.
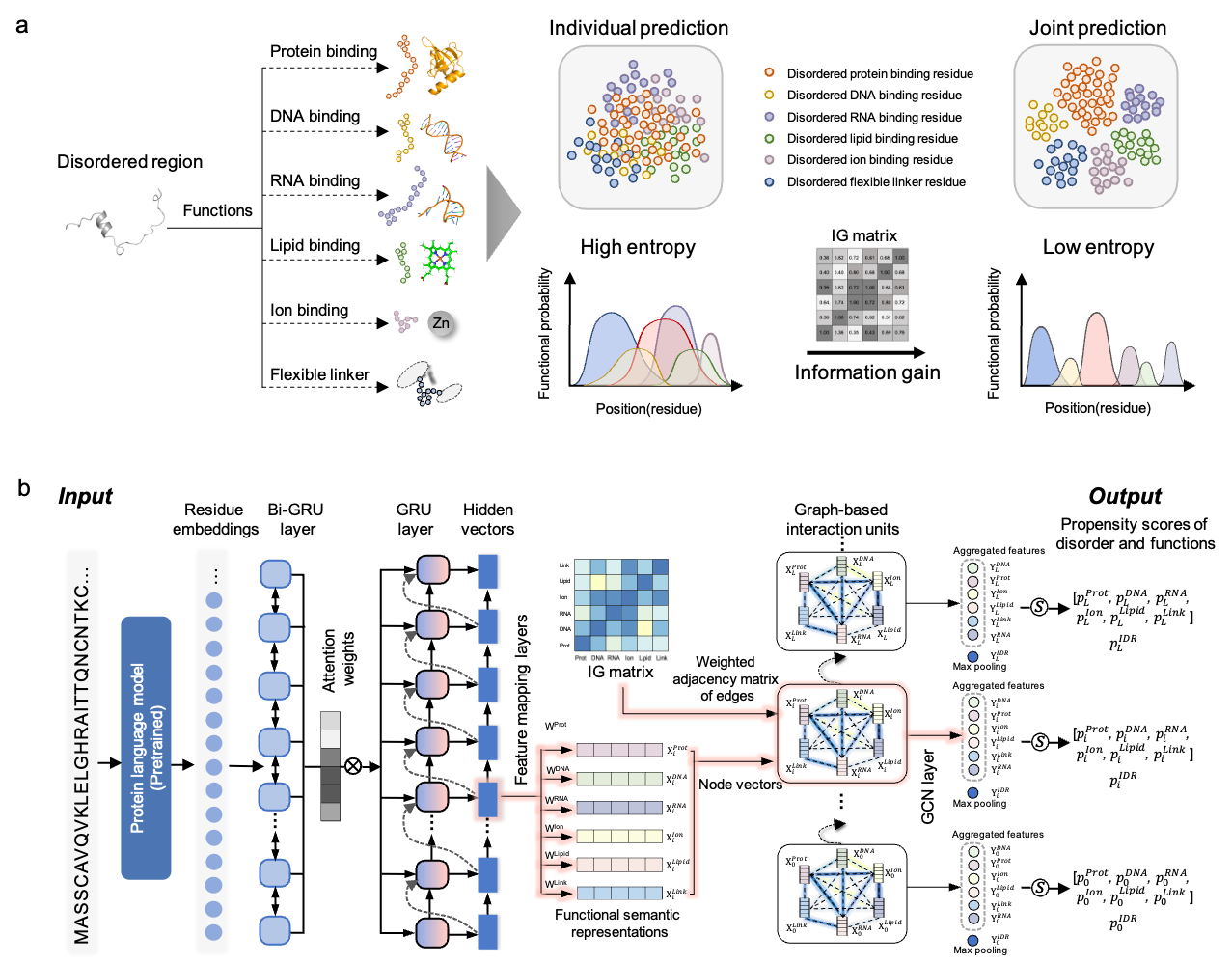
Figure.1 Schematic overview of DisoFLAG. (a) DisoFLAG provides predictions of six functions for intrinsically disordered regions in proteins. Joint prediction of the six functional regions results in a lower information entropy compared to individual prediction. The reduction in information entropy is known as information gain (IG), which reflects the correlation between different functions. High IG, strong correlation. (b) The graph-based interaction protein language model (GiPLM) architecture employed in DisoFLAG. The DisoFLAG predictor takes amino acid sequences as the only inputs and provides predictions of intrinsic disorder and six disordered functions for proteins, including protein-binding, DNA-binding, RNA-binding, ion-binding, lipid-binding, and flexible linker.
We acknowledge with thanks the following databases and softwares used in this server:
DisProt: database of intrinsically disordered proteins.
PDB: RCSB Protein Data Bank.
ProtTrans: Protein pre-trained language models.
Upon the usage the users are requested to use the following citation:
· Yihe Pang, Bin Liu. DisoFLAG: accurate prediction of protein intrinsic disorder and its functions using graph-based interaction protein language model. BMC Biology, 2024, 22: 3.