3 Tutorial
3.1 Introduction
The analysis of the structure of protein-containing complexes to extract binding residues is very important for the study of biology, pharmacology and bioinformatics. Although various databases or tools have been developed to summarize or analyze the interaction pairs in the complex, there are still cases where the results are incomplete or the presentation form is poor.
For example, for the widely used peptide-protein interaction database PepBDB[1], there are some problems, such as sequence residue deletion and insertion residue are not considered, and its last update is in March 2020. Besides, for ligand-protein interactions, the ligand contact tool LCT in FireDB[2] can be used to analyze the ligand binding residues of proteins contained in the complex, but the web tool is not friendly for batch analysis. Finally, for the protein-ligand interaction profiler PLIP[3,4], the insertion residue is also not considered in the analysis, and according to our application, it can not customize the threshold of binding residue atoms.
Therefore, PDB-BRE improved the above database or tools, fully considered the insertion residues in the complex structure, and supported the analysis of protein-protein interactions, peptide-protein interactions, DNA-protein interactions, RNA-protein interactions, NA hybrid-protein interactions and ligand-protein interactions. Compared with other databases or tools, PDB-BRE does not need to preprocess the PDB files in the RCSB PDB database before analysis, and will automatically extract binding residues from the complex according to the user-defined interaction distance threshold.
3.2 For protein-protein interaction
In the study of protein-protein interaction, PiSITE[5], as a web-based database of protein interaction sites, supports the provision of information about interaction sites of a protein from single PDB entry. However, PiSITE still has the following problems:
Due to a large number of changes in protein sequences and interaction sites, the protein-protein interaction data stored in the PiSITE database are out of date (the latest version is only updated to January 2019).
Considering that PiSITE is originally designed to identify binding residues in hub proteins based on multiple complexes, it does not support the analysis of interaction residues in a single complex.
Take the complex 1AFQ as an example, the binding residues involved in the protein chain C are recorded in PiSITE as shown in Figure 1 (screenshot from here). It is not difficult to see from Figure 1 that the sequence information is marked with different colors to indicate the number of times that each residue interacts with other protein chains in all PDB complexes (i.e. atomic distance is less than 4 Å, see PiSITE paper for details). However, PiSITE cannot provide the binding residues of proteins in a single complex, which may be of great significance for the study of single amino acid polymorphism.
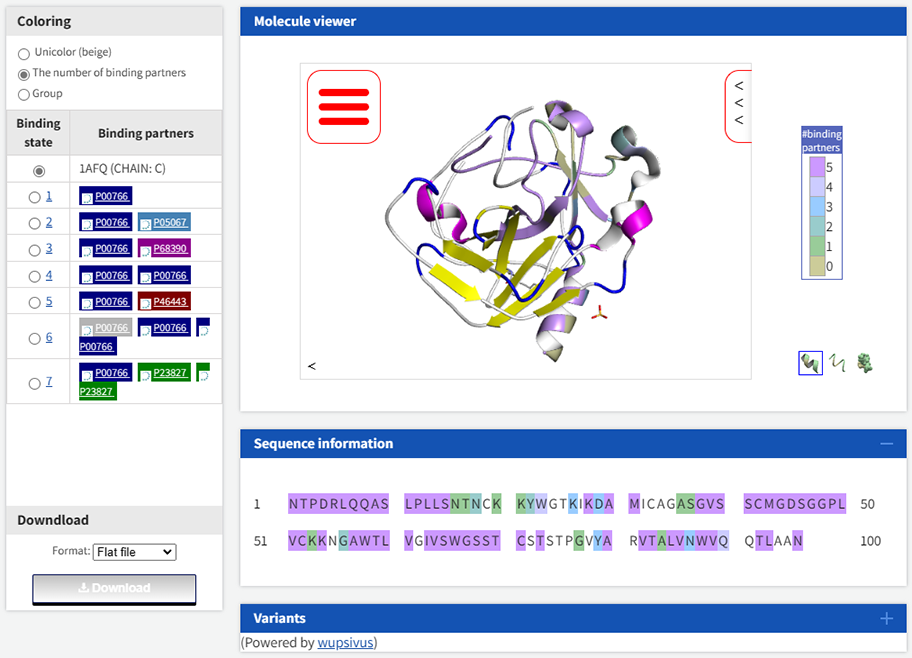
Therefore, PDB-BRE has made improvements to the problems existing in PiSITE. The binding residue information obtained from the analysis of chain C in 1AFQ by PDB-BRE is as follows. Specifically, the protein-protein interaction pairs in 1AFQ were analyzed by PDB-BRE-InterPair, and then the sequence label of chain C were extracted by PDB-BRE-PPISeqLabel. Among the result, the fourth column represents the new sequence label obtained after the protein chain is mapped to the UniProt database, and the eighth column represents the original sequence label of the protein chain. 1 indicates that the residue is a binding residue, and 0 indicates that the residue is an unbound residue.
InterPair_ID,UniProt ID,UniProt protein sequence,UniProt protein label,UniProt protein start,UniProt protein end,Complex protein sequence,Complex protein label,Mapping protein start,Mapping protein end
1afq_B_C,P00766,CGVPAIQPVLSGLSRIVNGEEAVPGSWPWQVSLQDKTGFHFCGGSLINENWVVTAAHCGVTTSDVVVAGEFDQGSSSEKIQKLKIAKVFKNSKYNSLTINNDITLLKLSTAASFSQTVSAVCLPSASDDFAAGTTCVTTGWGLTRYTNANTPDRLQQASLPLLSNTNCKKYWGTKIKDAMICAGASGVSSCMGDSGGPLVCKKNGAWTLVGIVSWGSSTCSTSTPGVYARVTALVNWVQQTLAAN,00000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000000111111111111111000000000000101100000011111111111111101000111101111001110100000010110111110011001,[150],[245],NTPDRLQQASLPLLSNTNCKKYWGTKIKDAMICAGASGVSSCMGDSGGPLVCKKNGAWTLVGIVSWGSSTCSTSTPGVYARVTALVNWVQQTLAAN,111111111111111000000000000101100000011111111111111101000111101111001110100000010110111110011001,[1],[96]
The overall comparison of PiSITE and PDB-BRE in protein-protein interactions is shown in Table 1.
Method |
Availability |
Distance threshold |
Presentation form |
Application |
|---|---|---|---|---|
PDB-BRE |
latest |
User customization |
Software package |
Based on single complex |
PiSITE |
Outdated |
4 Å |
Web server |
Based on multiple complexes |
3.3 For peptide-protein interaction
In the study of peptide-protein interaction, PepBDB collected the complex biological structures of peptide-mediated protein interactions in the Protein Data Bank[6] before March 2020. In PepBDB, the peptide lengths up to 50 residues. However, PepBDB still has the following problems:
Due to the update of the Protein Data Bank database, the peptide-protein interaction data stored in the PepBDB database are out of date (the latest version is only updated to March 2020).
Incomplete peptide or protein sequences are stored in the PepBDB database.Incomplete peptide or protein sequences are stored in the PepBDB database.
PepBDB does not fully consider the effect of insertion residues on peptide-protein interaction.
Take the complex 1THR as an example, the binding residues of the interaction between peptide chain L and protein chain H are recorded in PepBDB, as shown in Figure 2 (screenshot from here). As can be seen from Figure 2, the sequence length of the peptide chain L recorded in PepBDB is 27, while the real sequence length of L chain in PDB is 36, which is due to the loss of the head and tail of the sequence stored in PepBDB.
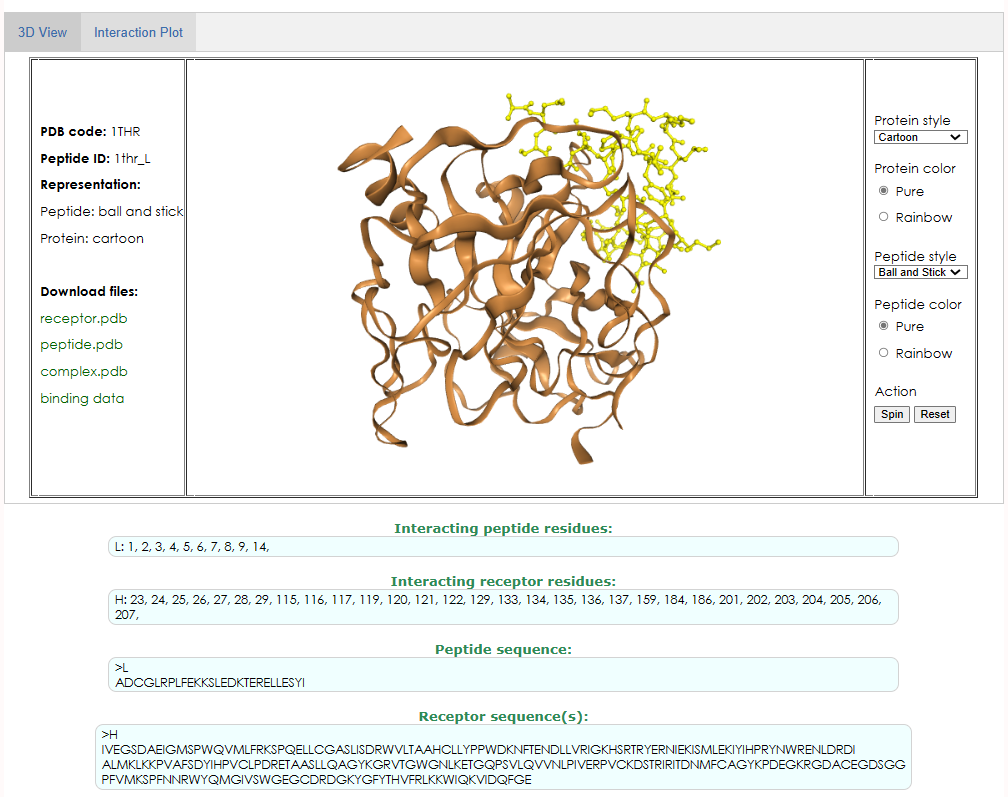
Beisides, in the study of proteins from different species, it has been found that there are certain connections between some sequences (for example, sequence evolution) or some important patterns (such as motif in some protein families). In order to better reflect this connection, to facilitate the discussion and comparison of the structure of different species, or to meet some external standard, people want to retain specific residue numbers rather than strictly incrementing them. For example, in the crystal structure 1THR, “1B” and “1A” residues are inserted before L chain residue 1 (A and B are insertion codes), as shown in Figure 3.
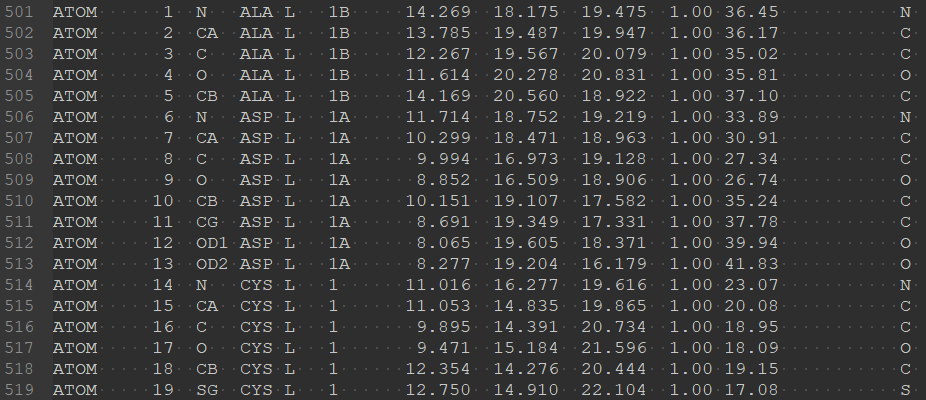
Unfortunately, PepBDB did not fully take into account the insertion code of the insertion residue when calculating the peptide-protein interaction. In addition, another widely used analytical tool, PLIP, also ignores the effect of insertion residues in peptide-protein interactions. Therefore, PDB-BRE has made improvements to the problems existing in PepBDB and PLIP. The binding residue information obtained from the analysis of chain L and chain H in 1THR by PDB-BRE is as follows.
InterPair_ID,Interacting peptide residues,Peptide sequence,Interacting receptor residues,Receptor sequence
1thr_L_H,"L:1B,1A,1,2,3,4,5,6,7,8,9,14,14A,14B,14C,14E,14F,14G,14I,14J",L:TFGSGEADCGLRPLFEKKSLEDKTERELLESYIDGR,"H:23,24,25,26,27,28,29,115,116,117,119,120,121,122,129C,133,134,135,136,137,159,184A,186D,201,202,203,204,204B,205,206,207",H:IVEGSDAEIGMSPWQVMLFRKSPQELLCGASLISDRWVLTAAHCLLYPPWDKNFTENDLLVRIGKHSRTRYERNIEKISMLEKIYIHPRYNWRENLDRDIALMKLKKPVAFSDYIHPVCLPDRETAASLLQAGYKGRVTGWGNLKETWTANVGKGQPSVLQVVNLPIVERPVCKDSTRIRITDNMFCAGYKPDEGKRGDACEGDSGGPFVMKSPFNNRWYQMGIVSWGEGCDRDGKYGFYTHVFRLKKWIQKVIDQFGE
The overall comparison of PepBDB and PDB-BRE in peptide-protein interactions is shown in Table 2.
Method |
Availability |
Cut-off Value of sequence length |
Distance threshold |
Presentation form |
Insert Residue |
|---|---|---|---|---|---|
PDB-BRE |
latest |
User customization |
User customization (Residual atomic distance) |
Software package |
Considered |
PepBDB |
Outdated |
50 |
5 Å (Residual atomic distance) |
Web server |
Not considered |
PLIP |
latest |
50 |
User customization (Non-covalent cooperation distance) |
Software package |
Not considered |
3.4 For DNA-protein interaction, NA-protein interaction and NA hybrid-protein interaction
In the study of NA-protein interaction(DNA-protein interaction, NA-protein interaction and NA hybrid-protein interaction), for example, Zhang et al. proposed NCBRPred[7] to predict nucleic acid binding residues in proteins, the data used are mainly YFK16-3.5 and YFK16-5 constructed by Yan et al. (For more information, see here)[8]. However, these data were obtained in September 2013. For the current analysis and study of NA-protein interaction, these data are incomplete and may lead to the loss of some important information. Besides, although PLIP can also analyze NA-protein interactions and extract corresponding protein-binding residues. However, PLIP also has the following problems:
In batch analysis, the processing steps of PLIP are more tedious.
PLIP does not fully consider the effect of insertion residues on NA-protein interaction.
Therefore, PDB-BRE has made improvements to the problems existing in current NA-protein interaction analysis tools. Take the complex 10MH as an example, the binding residue information obtained from the analysis of DNA-protein interaction in 10MH by PDB-BRE is as follows.
InterPair_ID,Interacting DNA residues,DNA sequence,Interacting receptor residues,Receptor sequence
10mh_B_A,"B:402,403,404,405,406,407,408,409,410,411,412",B:CCATGCGCTGAC,"A:44,86,87,90,122,123,126,209,234,236,237,239,240,255,256,257,258,260,261,294,296,297",A:MIEIKDKQLTGLRFIDLFAGLGGFRLALESCGAECVYSNEWDKYAQEVYEMNFGEKPEGDITQVNEKTIPDHDILCAGFPCQAFSISGKQKGFEDSRGTLFFDIARIVREKKPKVVFMENVKNFASHDNGNTLEVVKNTMNELDYSFHAKVLNALDYGIPQKRERIYMICFRNDLNIQNFQFPKPFELNTFVKDLLLPDSEVEHLVIDRKDLVMTNQEIEQTTPKTVRLGIVGKGGQGERIYSTRGIAITLSAYGGGIFAKTGGYLVNGKTRKLHPRECARVMGYPDSYKVHPSTSQAYKQFGNSVVINVLQYIAYNIGSSLNFKPY
10mh_C_A,"C:424,425,426,427,428,429,430",C:GTCAGCGCATGG,"A:78,79,80,81,82,85,86,87,88,89,97,119,120,121,162,163,164,165,226,228,237,240,242,249,250,251,252,253,254,255,256,303,304,305",A:MIEIKDKQLTGLRFIDLFAGLGGFRLALESCGAECVYSNEWDKYAQEVYEMNFGEKPEGDITQVNEKTIPDHDILCAGFPCQAFSISGKQKGFEDSRGTLFFDIARIVREKKPKVVFMENVKNFASHDNGNTLEVVKNTMNELDYSFHAKVLNALDYGIPQKRERIYMICFRNDLNIQNFQFPKPFELNTFVKDLLLPDSEVEHLVIDRKDLVMTNQEIEQTTPKTVRLGIVGKGGQGERIYSTRGIAITLSAYGGGIFAKTGGYLVNGKTRKLHPRECARVMGYPDSYKVHPSTSQAYKQFGNSVVINVLQYIAYNIGSSLNFKPY
3.5 For ligand-protein interaction
In the study of ligand-protein interaction, LCT is a tool for calculating the contact between protein and ligand atoms. Residues are considered in contact when one of his atoms is within a user-established Angstrom distance (default is 0.5) + Van der Waals radii.
However, LCT still has the following problems:
LCT needs to specify the ligand chain ID of the input complex structure before analysis, which is not convenient for batch analysis.
LCT does not fully consider the effect of insertion residues on ligand-protein interaction.
For multiple identical ligands that exist in the same chain, LCT cannot distinguish them in terms of results.
The distance criterion used by LCT is the user-specified threshold plus the van der Waals radius. When some studies only want to use the user-specified distance threshold, LCT’s calculation results are not acceptable.
Therefore, PDB-BRE has improved the problems existing in LCT so that it and LCT can complement each other in the study of ligand-protein interaction. Take the complex 1BZO as an example, the binding residue information obtained from the analysis of ligand in 1BZO by PDB-BRE is as follows.
InterPair_ID,Interacting ligand,Ligand formula,Interacting receptor residues,Receptor sequence
1bzo_ZN(151A)_A,A:151,ZN 2+,"A:44,61,68A,69,70,78,81,135",A:QDLTVKMTDLQTGKPVGTIELSQNKYGVVFIPELADLTPGMHGFHIHQNGSCASSEKDGKVVLGGAAGGHYDPEHTNKHGFPWTDDNHKGDLPALFVSANGLATNPVLAPRLTLKELKGHAIMIHAGGDNHSDMPKALGGGGARVACGVIQ
1bzo_CU(152A)_A,A:152,CU 2+,"A:44,46,61,116,118",A:QDLTVKMTDLQTGKPVGTIELSQNKYGVVFIPELADLTPGMHGFHIHQNGSCASSEKDGKVVLGGAAGGHYDPEHTNKHGFPWTDDNHKGDLPALFVSANGLATNPVLAPRLTLKELKGHAIMIHAGGDNHSDMPKALGGGGARVACGVIQ
1bzo_IUM(502A)_A,A:502,2(O2 U 2+),"A:68A,76",A:QDLTVKMTDLQTGKPVGTIELSQNKYGVVFIPELADLTPGMHGFHIHQNGSCASSEKDGKVVLGGAAGGHYDPEHTNKHGFPWTDDNHKGDLPALFVSANGLATNPVLAPRLTLKELKGHAIMIHAGGDNHSDMPKALGGGGARVACGVIQ
1bzo_IUM(559A)_A,A:559,2(O2 U 2+),"A:53B,53D,53E",A:QDLTVKMTDLQTGKPVGTIELSQNKYGVVFIPELADLTPGMHGFHIHQNGSCASSEKDGKVVLGGAAGGHYDPEHTNKHGFPWTDDNHKGDLPALFVSANGLATNPVLAPRLTLKELKGHAIMIHAGGDNHSDMPKALGGGGARVACGVIQ
The overall comparison of PepBDB and PDB-BRE in ligand-protein interactions is shown in Table 3.
Method |
Availability |
Cut-off Value of protein sequence length |
Distance threshold |
Presentation form |
Insert Residue |
|---|---|---|---|---|---|
PDB-BRE |
latest |
User customization |
User customization |
Software package (batch analysis) |
Considered |
LCT |
latest |
0 |
User customization + Van der Waals radii |
Web server (individual analysis) |
Not considered |
3.6 References
[1] Wen, Z., et al. PepBDB: a comprehensive structural database of biological peptide–protein interactions. Bioinformatics 2019;35(1):175-177.
[2] Maietta, P., et al. FireDB: a compendium of biological and pharmacologically relevant ligands. Nucleic acids research 2014;42(D1):D267-D272.
[3] Salentin, S., et al. PLIP: fully automated protein–ligand interaction profiler. Nucleic acids research 2015;43(W1):W443-W447.
[4] Adasme, M.F., et al. PLIP 2021: Expanding the scope of the protein–ligand interaction profiler to DNA and RNA. Nucleic acids research 2021;49(W1):W530-W534.
[5] Higurashi, M., Ishida, T. and Kinoshita, K. PiSite: a database of protein interaction sites using multiple binding states in the PDB. Nucleic acids research 2009;37(suppl_1):D360-D364.
[6] Rose, P.W., et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic acids research 2016:gkw1000.
[7] Zhang, J., Chen, Q. and Liu, B. NCBRPred: predicting nucleic acid binding residues in proteins based on multilabel learning. Briefings in bioinformatics 2021;22(5):bbaa397.
[8] Yan, J., Friedrich, S. and Kurgan, L. A comprehensive comparative review of sequence-based predictors of DNA-and RNA-binding residues. Briefings in bioinformatics 2016;17(1):88-105.