BioSeq-Analysis2.0: an updated platform for analyzing DNA, RNA, and protein sequences at sequence level and residue level based on machine learning approaches |
Established in 2017, the platform BioSeq-Analysis (1) is for the first time proposed to analyze various biological sequences at sequence-level based on machine learning approaches, which can do the following main steps automatically: feature extraction, predictor construction, and performance evaluation. BioSeq-Analysis has been increasingly and extensively applied in many areas of computational biology.
Moreover, many new and powerful predictors in the field of computational biology were developed by using the BioSeq-Analysis, such as iLearn (2), QSPred-FL (3), etc.
As shown in Fig. 1, there are two main important tasks in the field of biological sequence analysis, including residue-level analysis and sequence-level analysis.The aim of the residue-level analysis task is to study the properties of the residues, for instance protein-protein interaction site prediction (4), protein disordered region prediction (5), N6-Methyladenosine site prediction (6), etc, while the aim of the sequence-level analysis task is to investigate the structure and function characteristics of the entire sequences, such as enhancer identification (7, 8), protein remote homology detection and fold recognition (9-12), recombination spot identification (13, 14), DNA or RNA binding protein identification (15, 16), etc. All these biological sequence analysis tasks share three main processes, including feature extraction, predictor construction, and performance evaluation. The BioSeq-Analysis mainly focuses on analysing biological sequences at the sequence-level, meaning that the BioSeq-Analysis can be only applied to the sequence-level analysis tasks. Can we construct an intelligent tool to generate predictors for both residue-level and sequence-level analysis by automatically implementing all the three processes listed in Fig. 1? To answer this question, we have decided to publish an important updated platform called BioSeq-Analysis2.0. Compared with BioSeq-Analysis and other existing tools, BioSeq-Analysis 2.0 has the following new and novel functions and features:
I. 26 new feature extraction methods at the residue-level were added, of which 7 for DNA residues (17-21), 6 for RNA residues (17-19, 22), and 13 for amino acid residues (11, 17, 18, 23-32). To the best of our knowledge, BioSeq-Analysis2.0 is the first web server proposed to generate various residue-level feature extraction methods. As a result,BioSeq-Analysis2.0 covers a total of 26 modes at the residue-level and 56 modes at the sequence-level.
II. For the residue-level analysis tasks, a sliding window approach was applied to extract the information of the sequential neighboring residues, and a sequence labelling model Conditional Random Field (CRF) was added in BioSeq-Analysis2.0 so as to capture the global sequence order information of residues.
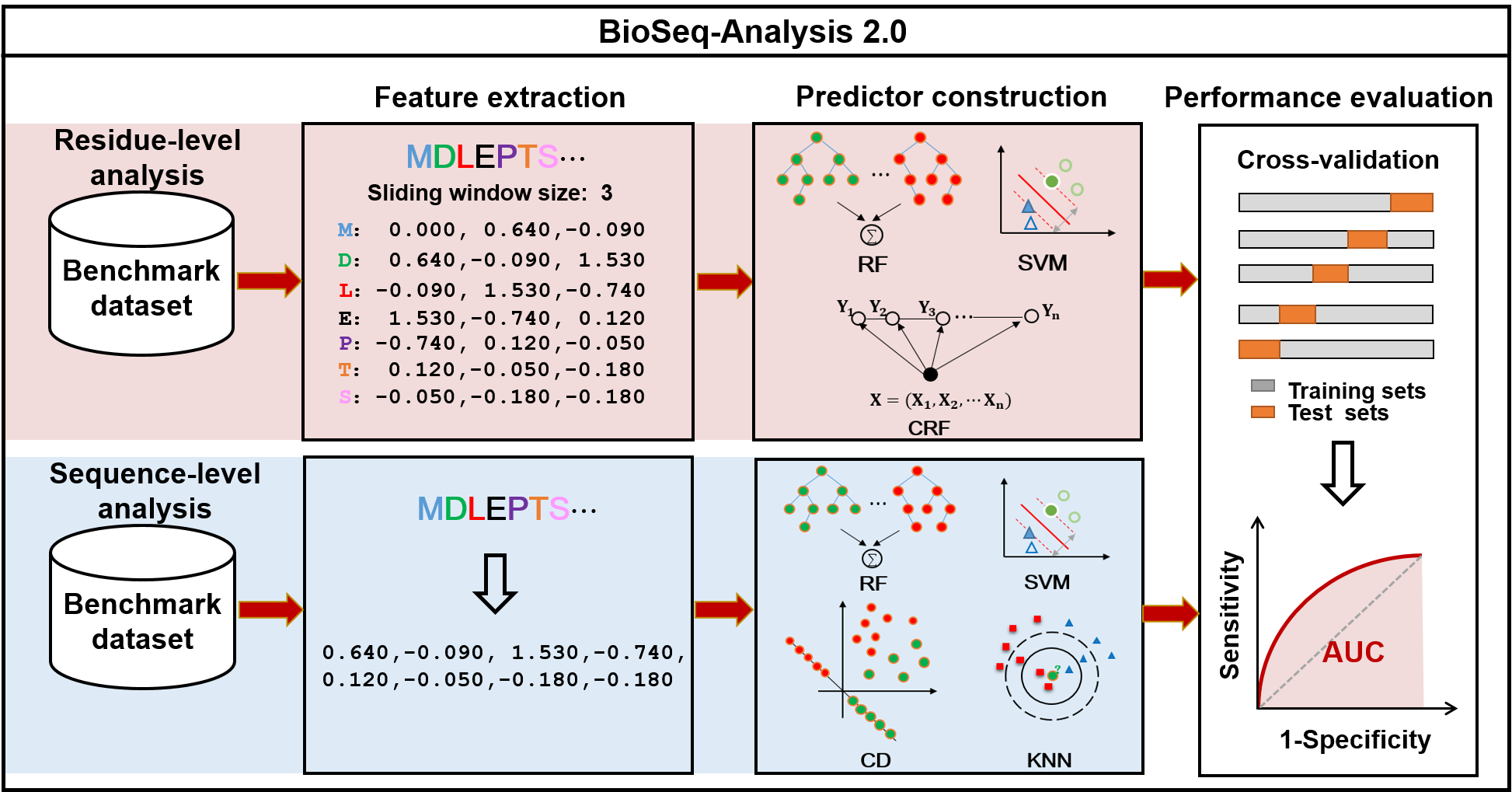
Fig. 1 The flowchart of BioSeq-Analysis2.0.
REFERENCES
1.Liu, B. (2017) BioSeq-Analysis: a platform for DNA, RNA and protein sequence analysis based on machine learning approaches. Briefings in Bioinformatics.
2.Chen, Z., Zhao, P., Li, F., Marquez-Lago, T.T., Leier, A., Revote, J., Zhu, Y., Powell, D.R., Akutsu, T., Webb, G.I. et al. (2019) iLearn: an integrated platform and meta-learner for feature engineering, machine-learning analysis and modeling of DNA, RNA and protein sequence data.Briefings in Bioinformatics. DOI: 10.1093/bib/bbz041.
3. Wei, L., Hu, J., Li, F., Song, J., Su, R. and Zou, Q. (2018) Comparative analysis and prediction of quorum-sensing peptides using feature representation learning and machine learning algorithms. Briefings in Bioinformatics. DOI: 10.1093/bib/bby107.
4. Bock, J.R. and Gough, D.A. (2001) Predicting protein-protein interactions from primary structure.Bioinformatics,17, 455-460.
5.Ishida, T. and Kinoshita, K.(2007) PrDOS: prediction of disordered protein regions from amino acid sequence.Nucleic Acids Research, 35, W460-464.
6.Zou, Q., Sr., Xing, P., Wei, L. and Liu, B. (2018) Gene2vec: Gene Subsequence Embedding for Prediction of Mammalian N6-Methyladenosine Sites from mRNA.RNA.
7.Liu, B., Fang, L.Y., Long, R., Lan, X. and Chou, K.C. (2016) iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition.Bioinformatics,32,362-369.
8.Liu, B., Li, K., Huang, D.S. and Chou, K.C.(2018) iEnhancer-EL: identifying enhancers and their strength with ensemble learning approach.Bioinformatics,34,3835-3842.
9.Chen, J.J., Guo, M.Y., Wang, X.L. and Liu, B. (2018) A comprehensive review and comparison of different computational methods for protein remote homology detection.Briefings in Bioinformatics,19,231-244.
10.Yan, K., Xu, Y., Fang, X.Z., Zheng, C.H. and Liu, B. (2017) Protein fold recognition based on sparse representation based classification.Artif Intell Med,79,1-8.
11.Liu, B., Zhang, D.Y., Xu, R.F., Xu, J.H., Wang, X.L., Chen, Q.C., Dong, Q.W. and Chou, K.C. (2014) Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection.Bioinformatics,30,472-479.
12.Chen, J.J., Guo, M.Y., Li, S.M. and Liu, B. (2017) ProtDec-LTR2.0: an improved method for protein remote homology detection by combining pseudo protein and supervised Learning to Rank.Bioinformatics,33,3473-3476.
13.Liu, B., Wang, X., Zou, Q., Dong, Q. and Chen, Q.(2013) Protein Remote Homology Detection by Combining Chou's Pseudo Amino Acid Composition and Profile-Based Protein Representation.Molecular Informatics,32,775-782.
14.Wang, R., Xu, Y. and Liu, B.(2016) Recombination spot identification Based on gapped k-mers.Sci Rep-Uk,6.
15.YYan, J., Friedrich, S. and Kurgan, L. (2016) A comprehensive comparative review of sequence-based predictors of DNA- and RNA-binding residues.Briefings in Bioinformatics,17,88-105.
16.Zhang, J. and Liu, B. (2017) PSFM-DBT: Identifying DNA-Binding Proteins by Combing Position Specific Frequency Matrix and Distance-Bigram Transformation.Int J Mol Sci,18.
17.Yoo, P.D., Zhou, B.B. and Zomaya, A.Y.(2008) Machine learning techniques for protein secondary structure prediction: An overview and evaluation. Curr Bioinform,3,74-86.
18.Doench, J.G., Fusi, N., Sullender, M., Hegde, M., Vaimberg, E.W., Donovan, K.F., Smith, I., Tothova, Z., Wilen, C., Orchard, R. et al.(2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol,34,184.
19.Chen, W., Zhang, X.T., Brooker, J., Lin, H., Zhang, L.Q. and Chou, K.C. (2015) PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions.Bioinformatics,31,119.
20.Friedel, M., Nikolajewa, S., Suhnel, J. and Wilhelm, T. (2009) DiProDB: a database for dinucleotide properties.Nucleic Acids Research,37,D37-D40.
21.Altschul, S., Madden, T., Schaffer, A., Zhang, J.H., Zhang, Z., Miller, W. and Lipman, D. (1998) Gapped BLAST and PSI-BLAST: A new generation of protein database search programs.Faseb J,12,A1326-A1326.
22.Hofacker, I.L., Fontana, W., Stadler, P.F., Bonhoeffer, L.S., Tacker, M. and Schuster, P. (1994) Fast Folding and Comparison of Rna Secondary Structures.Monatsh Chem,125,167-188.
23.Wang, J.T.L., Ma, Q., Shasha, D. and Wu, C.H. (2001) New techniques for extracting features from protein sequences.Ibm Syst J,40,426-441.
24.White, Seffens, G. and William. (1998) Using a neural network to backtranslate amino acid sequences.Electronic Journal of Biotechnology,1,17-18.
25.Lin, K., May, A.C.W. and Taylor, W.R.(2002) Amino acid encoding schemes from protein structure alignments: Multi-dimensional vectors to describe residue types.J Theor Biol,216,361-365.
26.Kawashima, S., Pokarowski, P., Pokarowska, M., Kolinski, A., Katayama, T. and Kanehisa, M.(2008) AAindex: amino acid index database, progress report 2008. Nucleic Acids Research, 36,D202-D205.
27.Cuff, J.A. and Barton, G.J.(2000) Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins, 40,502-511.
28.Heffernan, R., Paliwal, K., Lyons, J., Dehzangi, A., Sharma, A., Wang, J., Sattar, A., Yang, Y. and Zhou, Y. (2015) Improving prediction of secondary structure, local backbone angles, and solvent accessible surface area of proteins by iterative deep learning.Sci Rep,5,11476.
29.MO, D. (1978) A model of evolutionary change in proteins.Atlas Protein Seq.Struct.,5,89-99.
30.Henikoff, S. and Henikoff, J.G.(1992) Amino-Acid Substitution Matrices from Protein Blocks. P Natl Acad Sci USA,89, 10915-10919.
31.Altschul, S.F. and Koonin, E.V.(1998) Iterated profile searches with PSI-BLAST - a tool for discovery in protein databases. Trends Biochem Sci,23,444-447.
32.Glaser, F., Rosenberg, Y., Kessel, A., Pupko, T. and Ben-Tal, N. (2005) The ConSurf-HSSP database: The mapping of evolutionary conservation among homologs onto PDB structures.Proteins-Structure Function and Bioinformatics,58,610-617.