Pse-Analysis: a Python package for DNA/RNA and protein/peptide sequence analysis based on pseudo components and kernel methods |
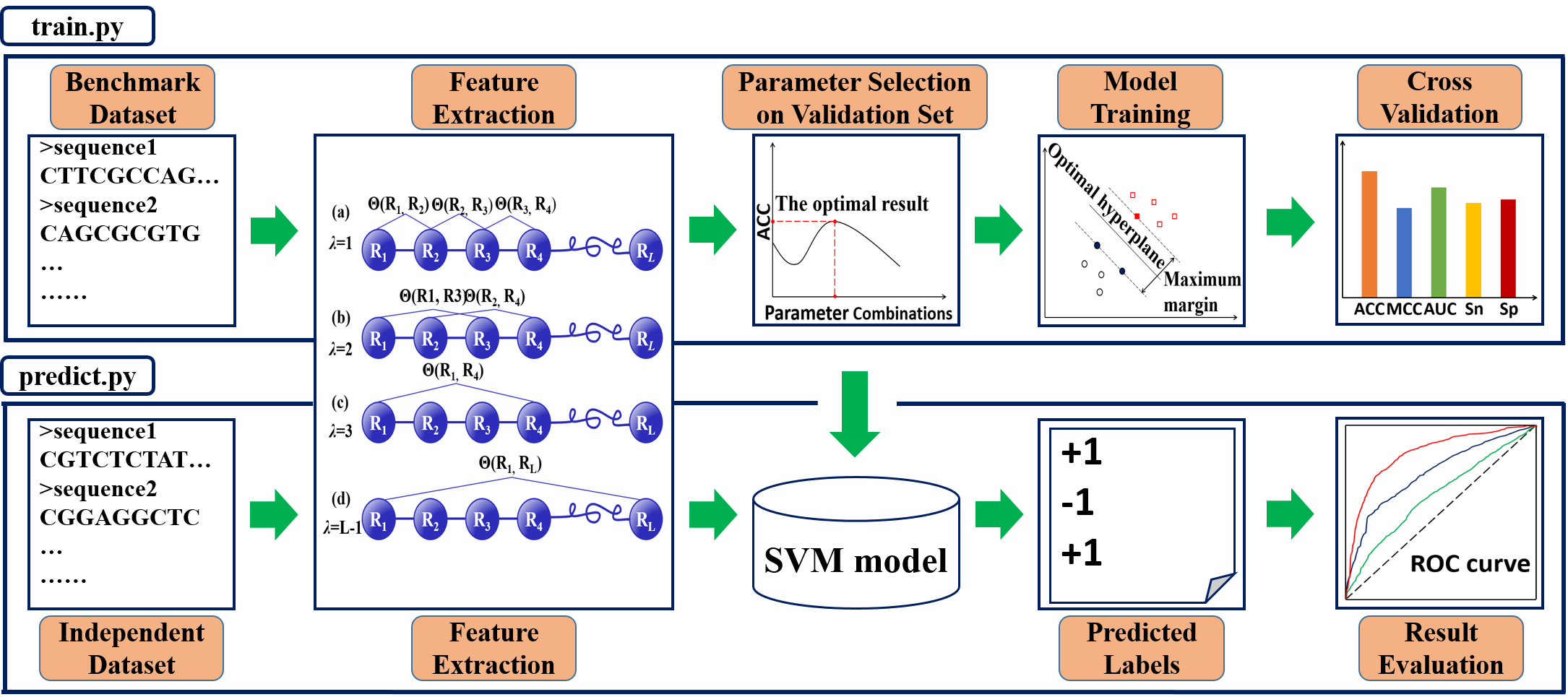
There are two scripts in Pse-Analysis package. One is “train.py” and the other is “predict.py”.
The “train.py” is used for training an SVM model. The procedure of “train.py” includes four steps: feature extraction, parameter selection, model training, and cross validation. In the feature extraction step, positive and negative datasets of DNA, RNA, or protein/peptide sequences are converted into fixed length feature vectors by using the PseKNC (1-4) or PseAAC (5-10), respectively. During the parameter selection step, the parameters of the feature extraction algorithm employed in the first step are automatically optimized on the validation procedure so as to reduce the risk of over-fitting problem. User-defined parameter settings (9), are also supported. The computational speed for the parameter selection is a bottleneck of the Pse-Analysis. In this regard, the multiprocessing technique is utilized to make the full usage of the available CPU cores. Experimental results on the nucleosome positioning prediction of C. elegans (11) show that the computing time can be significantly reduced by 6 folds when using 10 cores compared with that of the single core. In the model-training step, the feature vectors generated by the feature extraction algorithm with optimized parameters are fed into the SVM engine to train the classifiers. In this package, the LIBSVM package (12) is used to implement the SVM algorithm with RBF kernel. In the cross-validation step, three different strategies are provided, including k-fold cross validation, jackknife test, and independent dataset test. Five commonly used performance scores are calculated to evaluate the prediction quality from different angles, including accuracy (Acc), Mathew’s Correlation Coefficient (MCC), sensitivity (Sn), specificity (Sp), and area under ROC curve (AUC). Furthermore, the corresponding ROC (receiver operating characteristic) (13) curve is also provided and saved in a PNG file. Finally, a model file is generated, which will be used for the prediction in “predict.py” script.
In order for further evaluating the performance of the constructed predictors and for real-world application purpose, the “predict.py” script is provided to predict the query samples in an independent dataset. The results thus obtained are yielded in the output from Pse-Analysis along with the corresponding five scores mentioned above.
The flowchart of Pse-Analysis package
REFERENCES
1.Chen, W., et al. (2014b) PseKNC: a flexible web-server for generating pseudo K-tuple nucleotide composition Anal. Biochem, 456, 53-60.
2.Chen, W., et al. (2015b) PseKNC-General: a cross-platform package for generating various modes of pseudo nucleotide compositions Bioinformatics, 31, 119-120.
3.Liu, B., et al. (2015b) repDNA: a Python package to generate various modes of feature vectors for DNA sequences by incorporating user-defined physicochemical properties and sequence-order effects Bioinformatics, 31, 1307-1309.
4.Liu, B., et al. (2016b) repRNA: a web server for generating various feature vectors of RNA sequences Molecular Genetics and Genomics, 291, 473-481.
5.Cao, D.S., et al. (2013) propy: a tool to generate various modes of Chou's PseAAC Bioinformatics, 29, 960-962.
6.Chou, K.C. (2001) Prediction of protein cellular attributes using pseudo amino acid composition PROTEINS: Structure, Function, and Genetics (Erratum: ibid., 2001, Vol.44, 60), 43, 246-255.
7.Chou, K.C. (2009) Pseudo amino acid composition and its applications in bioinformatics, proteomics and system biology Current Proteomics, 6, 262-273.
8.Du, P., et al. (2014) PseAAC-General: Fast building various modes of general form of Chou's pseudo amino acid composition for large-scale protein datasets International Journal of Molecular Sciences, 15, 3495-3506.
9.Liu, B., et al. (2015c) Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences Nucleic Acids Res, 43, W65-W71.
10.Shen, H.B. and Chou, K.C. (2008) PseAAC: a flexible web-server for generating various kinds of protein pseudo amino acid composition Anal. Biochem, 373, 386-388.
11.Guo, S.H., et al. (2014) iNuc-PseKNC: a sequence-based predictor for predicting nucleosome positioning in genomes with pseudo k-tuple nucleotide composition Bioinformatics, 30, 1522-1529.
12.Chang, C.-C. and Lin, C.-J. (2011) LIBSVM: A library for support vector machines ACM Trans. Intell. Syst. Technol, 2, 1-27.
13.Fawcett, J.A. (2005) An Introduction to ROC Analysis Pattern Recognition Letters, 27, 861-874.
